
Todo el mundo recuerda a Stephen Hawking como uno de los grandes físicos de los últimos tiempos. Todos compartimos la imagen de un lucidísimo científico cuyas importantes restricciones físicas, como su incapacidad de caminar o de hablar, son compensadas por su inteligencia sin límites. De la misma forma, todos estamos familiarizados con el fenómeno viral Ice Bucket Challenge, consistente en grabarse en video vertiéndose un cubo de agua helada sobre la figura. Lo que quizá no todo el mundo sabe es que Stephen Hawking se desplazaba en silla de ruedas y la gente se vaciaba un cubo de agua helada ante una cámara por una misma razón: la Esclerosis Lateral Amiotrófica (ELA).
¿Qué es la Esclerosis Lateral Amiotrófica?
La ELA es una enfermedad neurodegenerativa caracterizada por una muerte neuronal progresiva, afectando principalmente a las neuronas motoras. Esta patología puede acabar desencadenando una parálisis total y la esperanza de vida suele ser de 2 a 3 años tras la aparición de los primeros síntomas.
La incidencia anual de la ELA en Europa es de entre 1 y 2 casos por cada 100.000 personas (Logroscino et al., 2009). En otras palabras, cada año aparecen en Europa entre 7000 y 15000 nuevos casos, el 90% de los cuales son esporádicos o no hereditarios (Valdmanis, Daoud, Dion & Rouleau, 2009). Por tanto, tan solo en el genoma del 10% restante pueden encontrarse mutaciones genéticas características de la ELA. La mayoría de pacientes tiene entre 40 y 70 años, siendo 55 la edad media de diagnóstico (The ALS Association, s.f.). La estadística muestra una incidencia mayor en los hombres que en las mujeres (Ralli, Lambiase, Artico, De Vincentiis & Greco, 2019).
¿Cómo se manifiesta la ELA?
Las manifestaciones clínicas de la ELA incluyen una combinación de signos derivados del mal funcionamiento de la neurona motora superior y de la neurona motora inferior. Estos son algunos de sus síntomas (National Institute of Neurological Disorders and Stroke, s.f.; The ALS Association, s.f.-b):
- Contracciones musculares en las extremidades o en la lengua.
- Fasciculaciones: pequeñas e involuntarias contracciones musculares, visibles bajo la piel y que no producen movimiento de miembros, debidas a descargas nerviosas espontáneas en grupos de fibras musculares esqueléticas (Colaboradores de Wikipedia, 2019).
- Calambres musculares.
- Rigidez muscular.
- Debilidad muscular (en las extremidades y en los músculos que posibilitan el habla).
- Dificultad para masticar y deglutir.
- Dificultad para respirar.
Las neuronas motoras conectan los músculos voluntarios con el sistema nervioso (véase Figura 1). Al degenerar estas neuronas, los músculos reciben una estimulación nerviosa pobre y se debilitan. En la mayoría de los casos, la enfermedad comienza afectando las extremidades. En otros casos, los primeros síntomas afectan al lenguaje y a la capacidad de deglutir los alimentos. En el 50% de los casos de ELA, los pacientes presentan además signos de demencia frontotemporal (Ralli, Lambiase, Artico, De Vincentiis & Greco, 2019).
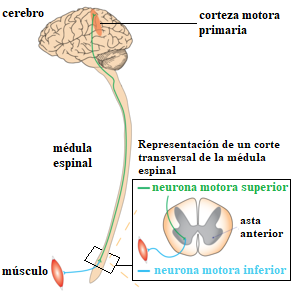
A medida que la enfermedad progresa, más neuronas motoras se van viendo afectadas y más partes del cuerpo pierden la movilidad voluntaria, hasta llegar a una parálisis total. Algunos músculos de la pared torácica como el diafragma pierden su movilidad, dificultando así la respiración (Ferguson & Elman, 2007; Hardiman, 2010; Lechtzin et al., 2018). Una de las diferencias de la Esclerosis Lateral Amiotrófica con respecto a otras enfermedades neurodegenerativas como el Alzheimer, es que los pacientes de esclerosis son plenamente conscientes de su estado físico, ya que las capacidades cognitivas se conservan. Este hecho hace que sea una de las enfermedades neurodegenerativas más duras psicológicamente.
La ELA no tiene un diagnóstico fácil, puesto que en la actualidad no existe ninguna prueba específica que lo permita. En el ámbito clínico, la identificación de la ELA pasa por una evaluación de los signos motores de una misma región del cuerpo. Según un estudio llevado a cabo por Paganoni et al. (2014), el tiempo medio requerido para hacer un diagnóstico definitivo es de entre 8 y 15 meses.
¿Cuál es la causa de la ELA?
La etiología es la parte de la medicina que estudia las causas de las enfermedades. La etiología de la ELA es muy compleja y por el momento no existe una respuesta definitiva. El origen de esta enfermedad neurodegenerativa es una incógnita para 9 de cada 10 casos, ya que tan solo los casos hereditarios (1 de cada 10) son identificables mediante un análisis del genoma. No obstante, se han elaborado algunas hipótesis para dar cuenta de la aparición de la enfermedad.
Hipótesis de los agregados
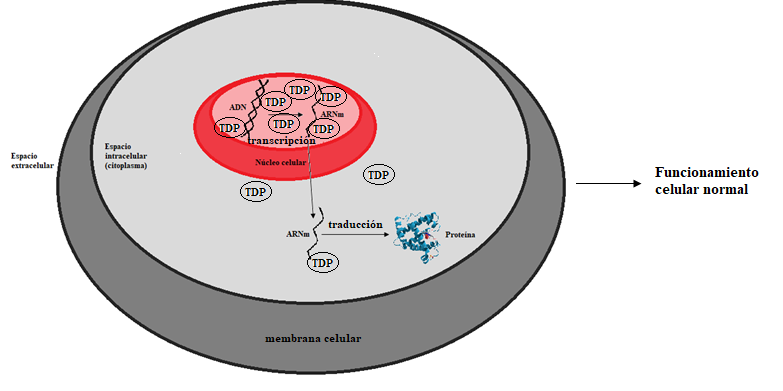
Algunos investigadores creen que una acumulación de agregados de proteínas en el interior de las células podría estar al origen de la neurodegeneración característica de la enfermedad. Antes de adentrarnos en esta hipótesis, recordemos brevemente el dogma central de la biología molecular a través de la Figura 2:

Volviendo a la enfermedad, Boeynaems (2017) explica en una conferencia de TEDx una interesante propuesta sobre los mecanismos moleculares de agregación de proteínas que subyacen a la ELA.
Según Boeynaems, la proteína responsable de la ELA es la TDP-43. TDP es una proteína necesaria para mantener el flujo de información genética de nuestras células (transcripción y traducción): el ADN, que se encuentra en el núcleo celular, debe ser transformado en ARN. La molécula de ARN sale entonces del núcleo y es leído por unas moléculas llamadas ribosomas, encargadas de sintetizar las proteínas a partir del código de ARN. Para que estos procesos puedan llevarse a cabo con normalidad, la concentración de TDP debe ser mayor en el núcleo que en el citoplasma. Una de las características de los pacientes de ELA es la perturbación de esta condición: las proteínas TDP se encuentran principalmente en el citoplasma, y una vez allí comienzan a agregarse y a formar placas que desencadenan la muerte neuronal. Un enfoque importante es el estudio del proceso que lleva a TDP a agregarse, ya que podría abrir vías terapéuticas para tratar la enfermedad.

Hipótesis de las mutaciones
Otros científicos apuestan por un problema genético: ciertas mutaciones en el genoma (modificaciones en el “manual de instrucciones”) dan lugar a la síntesis de proteínas anormales capaces de ejercer una nueva función tóxica para las células, y esto explicaría la neurodegeneración. Un ejemplo de mutación estrechamente relacionado con la ELA es la modificación de la Super Óxido Dismutasa (SOD). Esta mutación se encuentra en el 20% de los casos hereditarios de ELA (Sangwan & Eisenberg, 2016). Aún no está claro el lugar que ocupa esta proteína en la aparición de la enfermedad: se trata de una molécula que puede tener diferentes roles en función de la presencia o ausencia de mutación, así como en función de su localización celular (Pansarasa et al., 2018).
El estudio de los casos genéticos (comparando el genoma de pacientes de ELA hereditaria con personas sanas) permite localizar exactamente los genes clave implicados en la enfermedad. Uno de los genes que se han encontrado es el C9orf72 (Boeynaems, 2017). Esta mutación ha sido estudiada en células de levadura y en moscas. La mutación C9orf72 está relacionada con la perturbación del equilibrio de la proteína TDP-43 (explicado arriba), dando lugar a una acumulación citoplasmática con respecto al núcleo. Por lo tanto, las hipótesis no tienen por qué ser mutuamente excluyentes.
Hipótesis del sistema inmune
Además de la hipótesis de los “agregados” y de la hipótesis de las “mutaciones”, ciertas publicaciones atribuyen un rol central al sistema inmunitario (Ralli, Lambiase, Artico, De Vincentiis & Greco, 2019). Las células de nuestro sistema inmunitario se encargan de detectar y neutralizar las moléculas o los microbios susceptibles de ocasionarnos problemas que consiguen penetrar en nuestro organismo. En condiciones normales, estas células inmunitarias son especialistas en el reconocimiento de intrusos, y los diferencian perfectamente de las moléculas que forman parte de nuestro cuerpo o de los microorganismos que habitan pacíficamente nuestro organismo. Ciertas patologías consisten en una perturbación de este proceso de reconocimiento que da lugar a la identificación de moléculas endógenas (pertenecientes a nuestro organismo) como intrusos, y a su posterior destrucción. En estos casos hablamos de un mecanismo autoinmune. En cuanto a la ELA, no está claro cuál es la molécula endógena que podría ser reconocida por el sistema inmune desencadenando la defensa contra el propio organismo.
Lo que ahora sabes (y lo que no)
Ahora sabes por qué Stephen Hawking no podía caminar. La Esclerosis Lateral Amiotrófica es una enfermedad neurodegenerativa que afecta a las neuronas motoras, produciendo una atrofia muscular como consecuencia de su reducida estimulación. Los pacientes de ELA conservan sus capacidades cognitivas, pero la esperanza de vida es muy reducida a causa del gran deterioro físico. Hay varias hipótesis que tratan de explicar la etiología de la ELA: la hipótesis de los agregados, la hipótesis de las mutaciones, y la hipótesis del sistema inmune. Estas proposiciones no son contradictorias entre sí, pero no siempre es fácil establecer el vínculo entre los tres enfoques. Las tres teorías explican parte de la etiología de la ELA, pero no todas las incógnitas han sido resueltas. Aunque se han identificado moléculas que juegan un papel importante en la etiología de la ELA (proteínas como SOD1 o TDP-43; genes como C9orf72), aún hay muchas preguntas no resueltas relativas a la función de cada agente y a sus interacciones. En otras palabras, sabemos algunas cosas sobre ciertos procesos relacionados con la ELA, pero sabemos muy poco sobre la enfermedad en sí. Esto explica el hecho de que en la actualidad no exista ninguna cura y que nuestra mejor carta sea la mejora de la calidad de vida de los pacientes.
No son pocos los científicos que dedican su vida al estudio de esta enfermedad, ni son pocas las donaciones para que la investigación sobre la ELA sea posible. Pero una vez más, eso no es todo. En primer lugar, la ciencia no debe entenderse como una mera competición entre laboratorios. En lugar de una lucha entre diferentes doctrinas (por ejemplo, los defensores de la hipótesis del sistema inmune contra los defensores de la hipótesis de la agregación), debería fomentarse el intercambio de ideas, el debate, la síntesis de diferentes teorías.
En segundo lugar, la ciencia no debe ser un secreto entre los científicos y las entidades financieras. La comunidad científica no debe hablar un idioma diferente al que hablan la mayoría de los ciudadanos. Los avances científicos deberían ocupar el lugar privilegiado que ocupa el horóscopo en un periódico. La ciencia tiene un público demasiado limitado, el conocimiento se encuentra fragmentado, y la eficaz divulgación pseudocientífica obstaculiza su progreso. La comunidad científica debería preocuparse por transmitir sus mensajes a la totalidad de la sociedad, y por combatir el gran hermetismo que caracteriza a esta importante rama del conocimiento.
Fomentando una ciencia para todos, quizá algún día no será necesario explicar lo que había detrás de los cubos de agua helada.
Boeynaems, S. (2017, 5 enero). New insights in the molecular changes underlying ALS | Steven Boeynaems | TEDxUHasseltSalon [Archivo de vídeo]. Recuperado 13 octubre, 2019. Fuente
Colaboradores de Wikipedia. (2019, 12 octubre). Fasciculación. Recuperado 13 octubre, 2019. Fuente
Ferguson, T. A., & Elman, L. B. (2007). Neurorehabilitation Issues in Amyotrophic Lateral Sclerosis. Neurorehabilitation, 22(6), 409–416. Fuente
Hardiman, O. (2010). Management of respiratory symptoms in ALS. Journal of Neurology, 258(3), 359–365. Fuente
Lechtzin, N., Cudkowicz, M. E., De Carvalho, M., Genge, A., Hardiman, O., Mitsumoto, H., … Andrews, J. A. (2018). Respiratory measures in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, 19(5-6), 321–330. Fuente
Logroscino, G., Traynor, B. J., Hardiman, O., Chio, A., Mitchell, D., Swingler, R. J., … Beghi, E. (2009). Incidence of amyotrophic lateral sclerosis in Europe. Journal of Neurology, Neurosurgery & Psychiatry, 81(4), 385–390. Fuente
National Institute of Neurological Disorders and Stroke. (s.f.). Esclerosis lateral amiotrófica : National Institute of Neurological Disorders and Stroke (NINDS). Recuperado 13 octubre, 2019. Fuente
Pansarasa, O., Bordoni, M., Diamanti, L., Sproviero, D., Gagliardi, S., & Cereda, C. (2018). SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. International Journal of Molecular Sciences, 19(5), 1345. Fuente
Ralli, M., Lambiase, A., Artico, M., De Vincentiis, M., & Greco, A. (2019). Amyotrophic Lateral Sclerosis: Autoimmune Pathogenic Mechanisms, Clinical Features, and Therapeutic Perspectives. IMAJ, 21, 438–443. Fuente
Sangwan, S., & Eisenberg, D. S. (2016). Perspective on SOD1 mediated toxicity in Amyotrophic Lateral Sclerosis. Postepy Biochemii, 62(3), 362–369. Fuente
The ALS Association. (s.f.). Facts You Should Know. Recuperado 13 octubre, 2019. Fuente
Valdmanis, P. N., Daoud, H., Dion, P. A., & Rouleau, G. A. (2009). Recent advances in the genetics of amyotrophic lateral sclerosis. Current Neurology and Neuroscience Reports, 9(3), 198–205. Fuente
Wikipedia contributors. (2019, 10 octubre). Motor neuron disease. Recuperado 13 octubre, 2019. Fuente

