
En 1775, un ciclón tropical acabó con el 90% de la población de la isla de Pingelap. Tan solo unas 20 personas sobrevivieron. Entre uno de los supervivientes se encontraba el gobernante de la isla, quien además era portador de una enfermedad genética muy poco frecuente que le obligaba a ver el mundo en blanco y negro conocida como acromatopsia. El gobernante debió ser uno de los participantes más enardecidos en el proyecto de repoblar la isla, que a día de hoy cuenta con un 10% de la población con visión monocromática.

Este fenómeno es el resultado del efecto fundador, un interesante concepto evolutivo que puede explicar la aparición de características insólitas en los casos en los que un grupo reducido de individuos heterogéneo da lugar a una nueva población (Mosley, 2015).
Un siglo más tarde, en el mismo continente, tuvo lugar otro curioso caso de efecto fundador. Un médico americano llamado George Huntington describió los síntomas observados en una familia de ascendencia inglesa y bautizó con su propio nombre a la enfermedad neurodegenerativa. Se cree que los primeros casos tuvieron lugar en Europa del Norte, y dada la naturaleza hereditaria de la enfermedad se extendió rápidamente, especialmente en América. La población más grande conocida afectada por la enfermedad se encuentra en el estado venezolano mencionado previamente, Zulia. Los primeros habitantes llegaron a principios del siglo diecinueve, y al ser tan poco numerosos y haber algunos afectados, a lo largo del tiempo la enfermedad genética fue creciendo paralelamente a la población.
La enfermedad de Huntington
El Huntington es una enfermedad neurodegenerativa hereditaria que se caracteriza por la aparición de movimientos exagerados de las extremidades (movimientos coréicos), deterioro cognitivo y depresión. Se trata de una enfermedad genética desencadenada por una mutación situada en el cromosoma 4.
Síntomas
Las manifestaciones tempranas de la enfermedad aparecen en torno a los 35-40 años de edad, e incluyen movimientos indeseados de las extremidades que pueden dificultar la marcha, así como movimientos involuntarios faciales. Los problemas motores vienen acompañados de problemas cognitivos como dificultad para concentrarse y tomar decisiones. Es frecuente en los estadios tempranos observar problemas de irritabilidad y depresión (European Huntington Association, s.f.). A medida que la enfermedad progresa, aparecen otros síntomas motores como la lentitud en los movimientos o bradiquinesia, la rigidez muscular y los problemas de equilibrio.
En cuanto a los problemas cognitivos, los estadios avanzados de Huntington se caracterizan por una lentitud en el procesamiento de la información, una falta de conciencia de la propia enfermedad (síntoma conocido como anosognosia) y una incapacidad de realizar dos acciones simples a la vez. Asimismo, los pacientes muestran signos de apatía, depresión e impulsividad (European Huntington Association, s.f.).
El Huntington en cifras
La incidencia del Huntington es de 0,38 por cada 100.000 personas al año. Aplicado a la población española (46.934.632 personas) esto suponen 178 nuevos casos cada año. La esperanza de vida tras el diagnóstico es de 10 a 20 años (Ósmosis, 2019). Según una recopilación de meta-análisis realizada por Pringsheim et al. (2012), la prevalencia del Huntington (es decir, el número de casos estable en el tiempo, teniendo en cuenta que se trata de una enfermedad con una esperanza de vida limitada) es de 2,71 afectados por cada 100.000 personas.
Biologia del Huntington
La enfermedad del Huntington se caracteriza por la repetición anormal (36 veces o más) de una secuencia de ADN localizada en el cromosoma 4. Esta mutación tiene lugar en el gen que codifica la proteína huntingtina, y da lugar a una versión anormal de la misma que se relaciona con la aparición de los síntomas descritos previamente (aunque no está claro de qué manera). Se trata de una enfermedad autosómica dominante. ¿Qué quiere decir esto? Nuestro material genético está compuesto por 23 pares de cromosomas (es decir, somos organismos diploides): 22 pares de autosomas y un par de cromosomas sexuales que determinan, entre otras características, el sexo.

El cromosoma 4 en el que se localiza la mutación característica del Huntington es un autosoma. Cada par está compuesto por un ejemplar paterno y un ejemplar materno. Es decir, cada progenitor nos dota de una de sus dos versiones de cada cromosoma. Así, si la mutación se encuentra en uno de los cromosomas de uno de los antecesores (como suele ser el caso), hay un cincuenta por ciento de probabilidad de que un descendiente lo reciba. Algunas enfermedades exigen la presencia de dos versiones de la mutación (cada una proviniendo de uno de los antecesores). En el caso del Huntington no es así: la presencia de la mutación en uno de los cromosomas es suficiente para que la enfermedad se desarrolle. A esto se le conoce como enfermedad genética dominante (Osmosis, 2019).
La neurodegeneración tiene lugar principalmente en el núcleo caudado y el putamen, estructuras pertenecientes a los ganglios basales, regiones cerebrales relacionadas con el movimiento voluntario. El núcleo caudado y el putamen forman parte del estriado dorsal, y tienen un papel importante en la inhibición del movimiento. Esto puede explicar algunos de los síntomas principales de la enfermedad como los movimientos coréicos (puesto que hay una disminución de las células encargadas de inhibirlos).
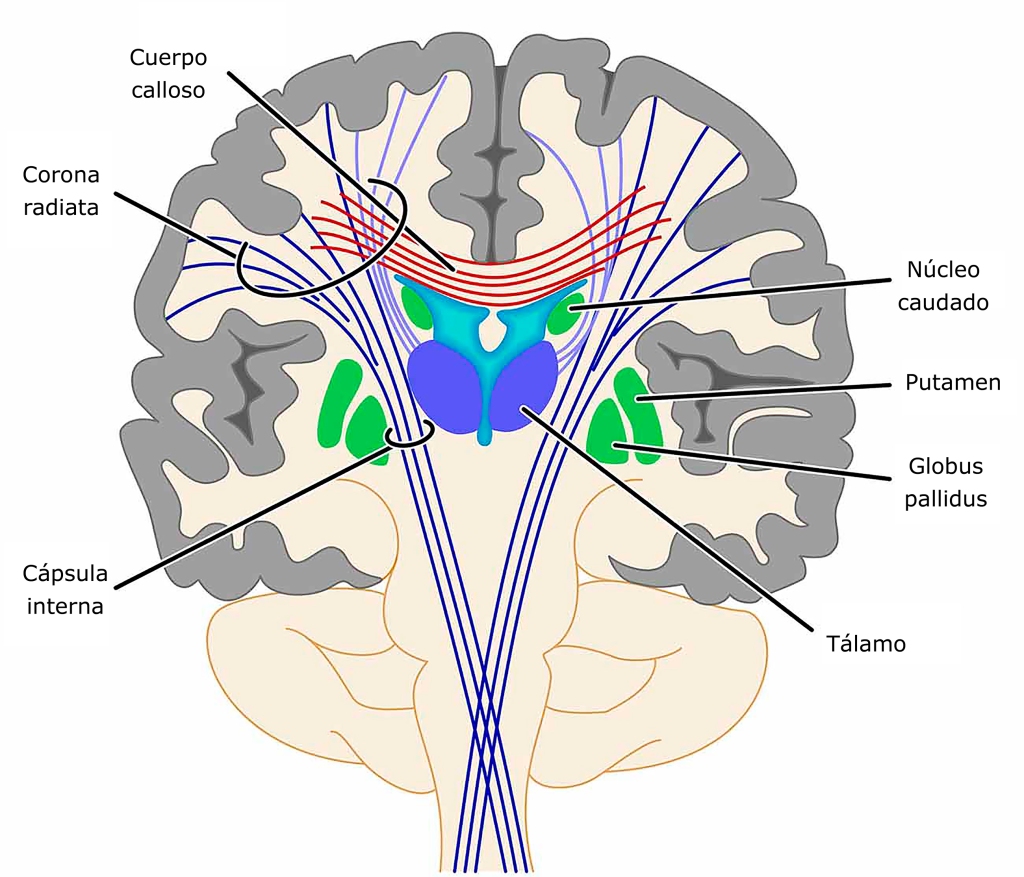
Cabe señalar que estos movimientos no pueden ser voluntariamente inhibidos, pero cesan durante las horas de sueño. La muerte neuronal es probablemente debida a una agregación proteica de la versión aberrante de la huntingtina. Las estructuras cerebrales afectadas en el Huntington se caracterizan además por tener una actividad neurotransmisora perturbada (es decir, la actividad de las moléculas encargadas de establecer la comunicación entre las neuronas se ve afectada). Concretamente, la actividad gabaérgica y acetilcolinérgica se ve disminuída, mientras que la actividad dopaminérgica se ve incrementada (Osmosis, 2019).
Las repeticiones de las secuencias de ADN no solo afectan a la huntingtina, sino que también se relacionan con problemas de la replicación del ADN (proceso mediante el cual la célula genera una copia idéntica del ADN para que cada célula hija reciba un ejemplar tras la división celular). En efecto, la proteína encargada de sintetizar la copia de ADN a partir de la secuencia de ADN que le sirve como modelo puede perder el hilo y crear más repeticiones de las que había originalmente. En otras palabras, a medida que la enfermedad va siendo heredada, el número de repeticiones puede incrementar en la descendencia. Esto tiene a su vez un efecto sobre la aparición de los síntomas debido al efecto de “anticipación”: cada nueva generación que hereda la mutación muestra los signos en estadios más tempranos. Este fenómeno de expansión de repeticiones ocurre mayoritariamente en las células sexuales masculinas, por lo que es más frecuente observarlas en los casos en los que el ascendiente paterno posee la mutación.
La prueba genética que permite predecir la aparición o diagnosticar la enfermedad de Huntington consiste en contar el número de repeticiones, puesto que correlaciona con la aparición de los síntomas (Osmosis, 2019).
Lo que sabes y lo que no
El Huntington es una enfermedad neurodegenerativa con un importante componente genético. La muerte neuronal tiene lugar en el estriado dorsal, una estructura implicada en la regulación del movimiento voluntario. Un componente molecular que podría estar implicado en la degeneración celular es la huntingtina, la proteína mutante que resulta de la mutación genética característica del huntington: número anormal de repeticiones de una secuencia de ADN. Aún no está claro cómo la huntingtina provoca la muerte neuronal, pero una hipótesis es que la proteína resultante de la mutación posee componentes que pueden interactuar de forma afín y formar agregados proteicos. Dichos agregados resultan tóxicos para las células y podrían inducir la neurodegeneración.
European Huntington Association. (s.f.). What is Huntington’s Disease? – European Huntington Association. Recuperado 22 diciembre, 2019. Fuente
Mosley, M. (2015, 28 septiembre). La isla llena de gente que sólo puede ver en blanco y negro. Recuperado 22 diciembre, 2019. Fuente
Osmosis. (2019, 7 mayo). Huntington disease - causes, symptoms, diagnosis, treatment & pathology [Archivo de vídeo ]. Recuperado 22 diciembre, 2019. Fuente
Pringsheim, T., Wiltshire, K., Day, L., Dykeman, J., Steeves, T., & Jette, N. (2012). The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Movement Disorders, 27(9), 1083–1091. Fuente

